In health, our cells work as fine-tuned machines following precise instructions, as computers execute software, for example. Which genes, for how long they are switched on and how potent they are, define the normal behavior of cells largely. Yet, it is very difficult to study diseases if we don’t know precisely how cells communicate with one another and how this gene expression is spread in a tissue. With today’s technology, we get a detailed report from each cell about its gene expression, but we cannot record where this cell was in the tissue.
Our goal is to use these mature methods in combination with methods that do record the position but have other disadvantages and develop algorithms that attempt to reconstruct computationally where the cells were situated inside the tissue in the first place. This way, we’ll be able to better understand how diseases get established and how they progress for the ultimate aim of developing personalized treatments in the future.
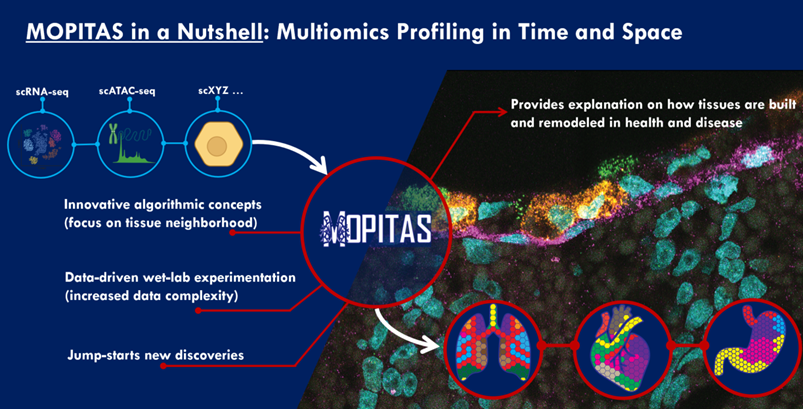
MOPITAS aims to develop experimental techniques and new algorithms to track single cells in space and time by combining spatial transcriptomics (ST) and single-cell RNA sequencing (scRNA-seq). The research could lead to scientific breakthroughs in tissue organization and its disruption in disease, as well as advance the understanding of tissue evolution in general.
The project combines generation of novel data with the development of novel and highly powerful computational methods to address fundamental scientific questions relating to the influence and interplay of cellular context and gene expression in healthy and diseased tissue.
This ambitious project can be summarized in the following goals that will advance our understanding of tissue organization:
MOPITAS aims to:
- Development of complex algorithms for the spatial layout of single cells in space according to a ST reference. Existing data will be utilized to create artificial data to thoroughly evaluate and test the algorithmic approaches.
- Benefitting from the learned neighborhood relationships of tissues, contrast learning can identify local neighborhood in tissue and create the assembly of reference free tissue maps.
- Using a joint spatial layout of transcriptomics and epigenomic cell profiles allows us to determine regulatory mechanisms and changes in gene-regulatory programs in health and disease throughout developmental and evolutionary time.
- Moving beyond the direct comparison of individual spatial multi-omes, we strive for a model of semantic spatial integration, i.e. we directly map cells across diverse tissue samples across individuals and across developmental and evolutionary time.